不眠症治療の最終ゴールとは
監修 秋田大学大学院医学系研究科 医学専攻 病態制御医学系 精神科学講座 三島 和夫 先生*
*資材ご監修時のお肩書で掲載させていただいております。
不眠症は夜間の不眠症状に加えて、不眠に起因する日中の精神・身体機能の低下(QOL障害)が生じる疾患として定義されています 1)
睡眠障害国際分類 第3版 改訂版(ICSD‑3-TR)における不眠症の診断基準 2)

そのため、不眠症の治療においては夜間症状(不眠症状)のみならず、適切な治療を提供することでQOLを含む日中機能の向上を目指すことが重要です 4)。
ク−ビビック錠の承認時評価試験においては、日中機能の評価指標*1としてIDSIQ*2が組み込まれています。
*2 IDSIQは、睡眠の質を評価することを目的にピッツバーグ大学にて作成されたDaytime Insomnia Symptom Scale 5)を再構成し、イドルシア社が新たに開発した評価ツールです。本評価ツールはFDAのPROガイダンスに従い開発され、不眠症評価指標として信頼できる、と評価されています 6)。
日中機能を評価する指標 〜IDSIQとは〜
- 14の質問[Sleepiness(4項目)、Alert/Cognition(6項目)、Mood(4項目)の3つの領域]により構成され、各質問に対し0〜10の11段階のスケールで評価し、それぞれの領域における合計スコアと、14項目の合計スコアによる評価を行います 6)。
- 臨床試験においては、タブレット型端末を利用し、Patient Reported Outcome(PRO:患者報告アウトカム)を実施しました。

臨床成績
国内第Ⅲ相試験(ID-078A304試験)
社内資料:日本人不眠症患者を対象とした国内第Ⅲ相試験[ID-078A304試験](承認時評価資料)
Uchimura N, et al.: Sleep Med. 2024; 122: 27-34.
(本研究はNxera Pharma Japanの資金により行われた。著者にNxera Pharma Japan社員が含まれる。
著者にNxera Pharma Japanよりコンサルタント料等を受領している者が含まれる。)
試験概要
- 目的
-
日本人不眠症患者におけるクービビックの有効性及び安全性を評価する。
- 試験デザイン
-
多施設共同、ランダム化、二重盲検、プラセボ対照、並行群間比較試験
- 対象
-
不眠症患者490例
<主な登録基準>
・18歳以上、BMIが30.0kg/m2未満の男女
・DSM-5に基づき不眠障害と診断された患者
・初回スクリーニング(来院1回目)前の12週以上にわたって週3夜以上、自己報告による以下のすべての症状がある患者
(a)主観的睡眠潜時(sLSO)30分以上 (b)主観的中途覚醒時間(sWASO)30分以上
(c)主観的総睡眠時間(sTST)6.5時間以下
・来院1回目の不眠重症度指数(ISI)スコアが15以上の患者
・来院1回目で普段の就床時刻(中央値)が20:30~00:30、就床時間(中央値)が6時間から9時間と報告した患者
・プラセボrun-in期(来院2回目)の直前7日間記入した睡眠日誌において、3夜以上で以下のすべてに該当する患者
(a)主観的睡眠潜時(sLSO)30分以上 (b)主観的中途覚醒時間(sWASO)30分以上
(c)主観的総睡眠時間(sTST)6.5時間以下
・来院2回目の直前7日間記入した睡眠日誌において、普段の就床時刻(中央値)が20:30~00:30、就床時間(中央値)が6時間から9時間と報告した患者
・ランダム化(来院3回目)の直前7日間のうち6日以上プラセボを服用し、睡眠日誌においてプラセボ服用日のうち3夜以上で以下のすべてに該当する患者
(a)主観的睡眠潜時(sLSO)30分以上 (b)主観的中途覚醒時間(sWASO)30分以上
(c)主観的総睡眠時間(sTST)6.5時間以下
・来院3回目の直前7日間記入した睡眠日誌において、普段の就床時刻(中央値)が20:30~00:30、就床時間(中央値)が6時間から9時間と報告した患者
・睡眠関連呼吸障害(慢性閉塞性肺疾患等)、睡眠時無呼吸症候群の既往歴がなく、合併していない患者
・周期性四肢運動障害、レストレスレッグス症候群、概日リズム睡眠障害、レム睡眠行動障害、ナルコレプシーをいずれも合併していない患者、精神疾患(不安障害、大うつ病、双極性障害、統合失調症、強迫性障害 等)を有していない患者
- 方法
-
本試験はスクリーニング期(7~18日間)、単盲検のプラセボrun-in期(14~20日間)、二重盲検期(28日間)、プラセボrun-out期(7日間)及びフォローアップ期(23日間)で構成された。
プラセボrun-in期の後、クービビック25mg群、50mg群又はプラセボ群に1:1:1の比でランダムに割付け、二重盲検下で1日1回就寝前に4週間経口投与した。その後、単盲検にてプラセボを1日1回就寝前に7日間経口投与した。
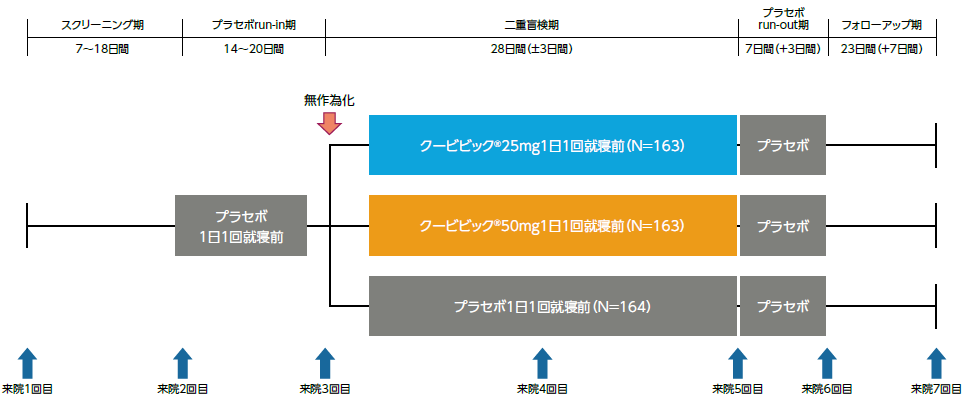
- 評価項目
-
主要評価項目(有効性):検証的解析項目
・4週時における主観的総睡眠時間(sTST)ならびに主観的睡眠潜時(sLSO)のベースラインからの変化量のクービビック50mg群とプラセボ群の比較
副次評価項目(有効性):
・4週時における主観的総睡眠時間(sTST)ならびに主観的睡眠潜時(sLSO)のベースラインからの変化量のクービビック25mg群とプラセボ群の比較
探索的評価項目:
・2週時における主観的総睡眠時間(sTST)のベースラインからの変化量のクービビック25mg群、50mg群とプラセボ群の比較
・2週時における主観的睡眠潜時(sLSO)のベースラインからの変化量のクービビック25mg群、50mg群とプラセボ群の比較
・4週時における主観的中途覚醒時間(sWASO)のベースラインからの変化量(事後解析)
・4週時における主観的睡眠効率[100×主観的総睡眠時間(sTST)/眠ろうとし始めた時から最後に目が覚めた時までの時間]のベースラインからの変化量
・4週時における中途覚醒回数のベースラインからの変化量
・4週時におけるIDSIQの各スコアのベースラインからの変化量 等
安全性評価項目:
有害事象、バイタルサイン、臨床検査、心電図、C-SSRS、反跳性不眠、退薬症候[ベンゾジアゼピンによる離脱症状に関する質問票(BWSQ)]、日中の眠気[エプワース眠気尺度日本語版(JESS)]、翌日の持ち越し効果[WAIS-IV知能検査(符号)] 等
- 解析計画
-
解析対象集団:
有効性の解析対象集団は、ランダム化され少なくとも1回治験薬を投与されたすべての患者(FAS)、安全性の解析対象集団は治験薬を1回以上投与されたすべての患者とした。
主要評価項目(2週時は探索的評価項目):
主観的総睡眠時間(sTST)及び主観的睡眠潜時(sLSO)のベースラインからの変化量について、それぞれのベースライン値、投与群、年齢区分(65歳未満、65歳以上)、評価時点(2週時、4週時)を固定効果、被験者を変量効果、投与群と評価時点及びベースライン値と評価時点を交互作用項とした線形混合効果モデルを用いて解析した。線形混合効果モデルは、無構造共分散行列を基本として測定間の相関をモデル化し、対比を用いてプラセボ群とクービビック50mg群を比較した。
検定の多重性による第1種の過誤の増大を防ぐため、階層的閉手順により4週時における主観的総睡眠時間(sTST)についてプラセボ群とクービビック50mg群との比較を行い、両側有意水準0.05で有意差が認められた場合、4週時における主観的睡眠潜時(sLSO)についてプラセボ群とクービビック50mg群を比較した。
なお、主観的総睡眠時間(sTST)及び主観的睡眠潜時(sLSO)とも、評価時点(2週時、4週時)の週平均は、評価日及びその直前の6日間(少なくとも3日間)における平均値とし、1週間のデータが3日未満であった場合、その週平均は欠測とし、主要解析においては欠測値の補完は行わず、線形混合効果モデルを適用した。
副次評価項目:
主要評価項目である4週時における主観的睡眠潜時(sLSO)の帰無仮説が棄却された場合、検定の多重性による第1種の過誤の増大を防ぐため、階層的閉手順により4週時における主観的総睡眠時間(sTST)についてプラセボ群とクービビック25mg群との比較を行い、両側有意水準0.05で有意差が認められた場合、4週時における主観的睡眠潜時(sLSO)についてプラセボ群とクービビック25mg群を比較した。主要評価項目の解析で用いるモデルを用い、対比によりプラセボ群とクービビック25mg群を比較した。
探索的評価項目:
連続量については要約統計量(例数、平均、標準偏差、中央値、最小値、最大値)を算出し、分類データについては例数とその割合、必要に応じて発現割合の95%CIを算出した。サブグループ解析:
主要評価項目及び副次評価項目について、年齢区分(65歳未満、65歳以上)、性別(男性、女性)、併存疾患(あり、なし)によりサブグループ解析を実施することを事前に計画した。
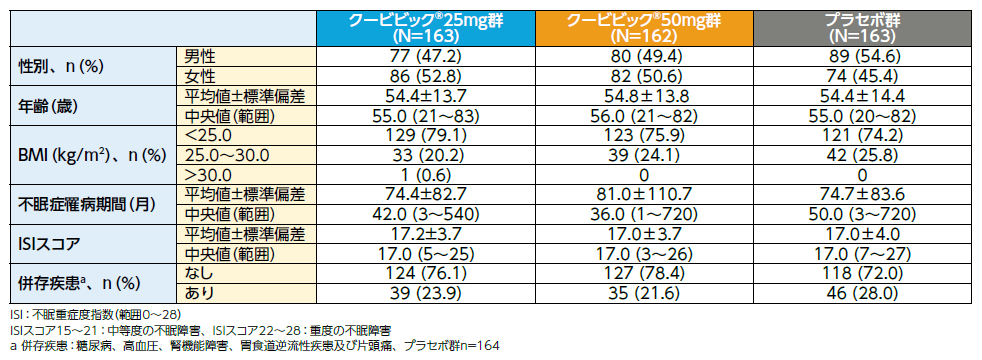
患者背景(FAS)
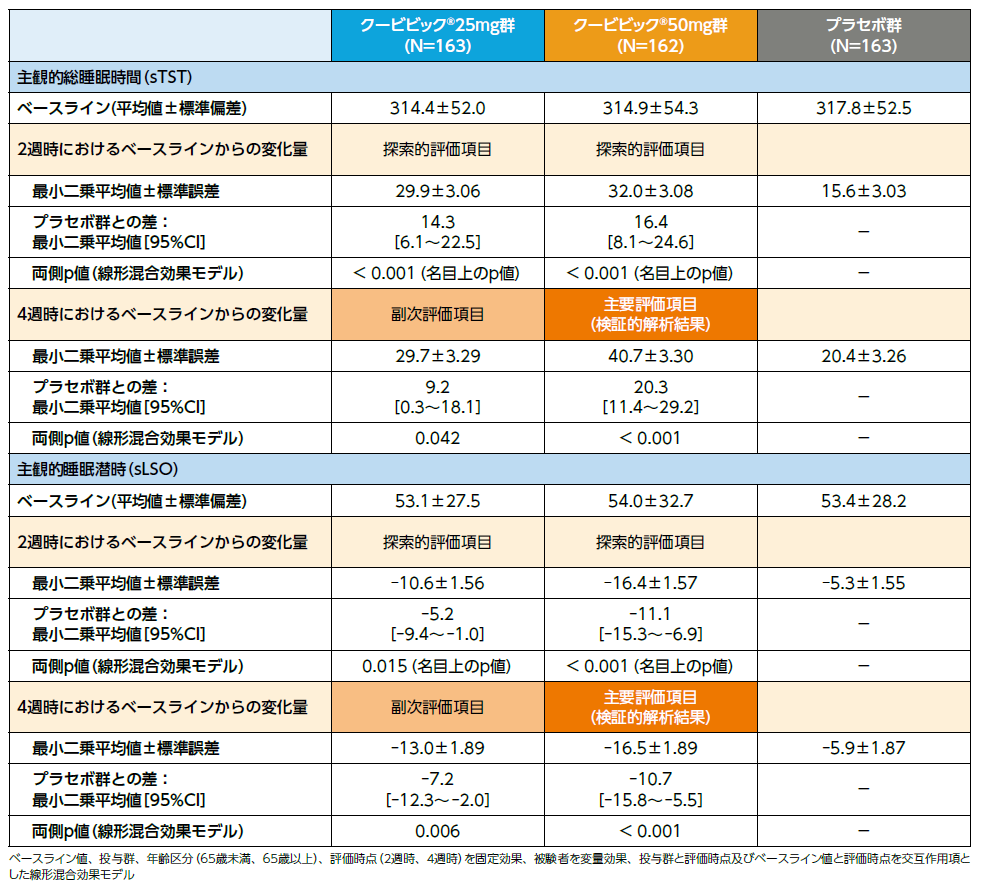
有効性のまとめ:主要評価項目(検証的解析結果)、副次評価項目、探索的評価項目
第1の主要評価項目である4週時における主観的総睡眠時間(sTST)のベースラインからの変化量のクービビック50mg群とプラセボ群との差(最小二乗平均値)は20.3分[95%CI:11.4〜29.2]であり、sTSTはプラセボ群と比較してクービビック50mg群で有意に延長しました(p<0.001、線形混合効果モデル)(検証的解析結果)。副次評価項目である4週時におけるsTSTのベースラインからの変化量のクービビック25mg群とプラセボ群との差(最小二乗平均値)は9.2分[95%CI:0.3〜18.1]であり、sTSTはプラセボ群と比較してクービビック25mg群で有意に延長しました(p=0.042、線形混合効果モデル)。
第2の主要評価項目である4週時における主観的睡眠潜時(sLSO)のベースラインからの変化量のクービビック50mg群とプラセボ群との差(最小二乗平均値)は-10.7分[95%CI:-15.8〜-5.5]であり、sLSOはプラセボ群と比較してクービビック50mg群で有意に短縮しました(p<0.001、線形混合効果モデル)(検証的解析結果)。副次評価項目である4週時におけるsLSOのベースラインからの変化量のクービビック25mg群とプラセボ群との差(最小二乗平均値)は-7.2分[95%CI:-12.3〜-2.0]であり、sLSOはプラセボ群と比較してクービビック25mg群で有意に短縮しました(p=0.006、線形混合効果モデル)。
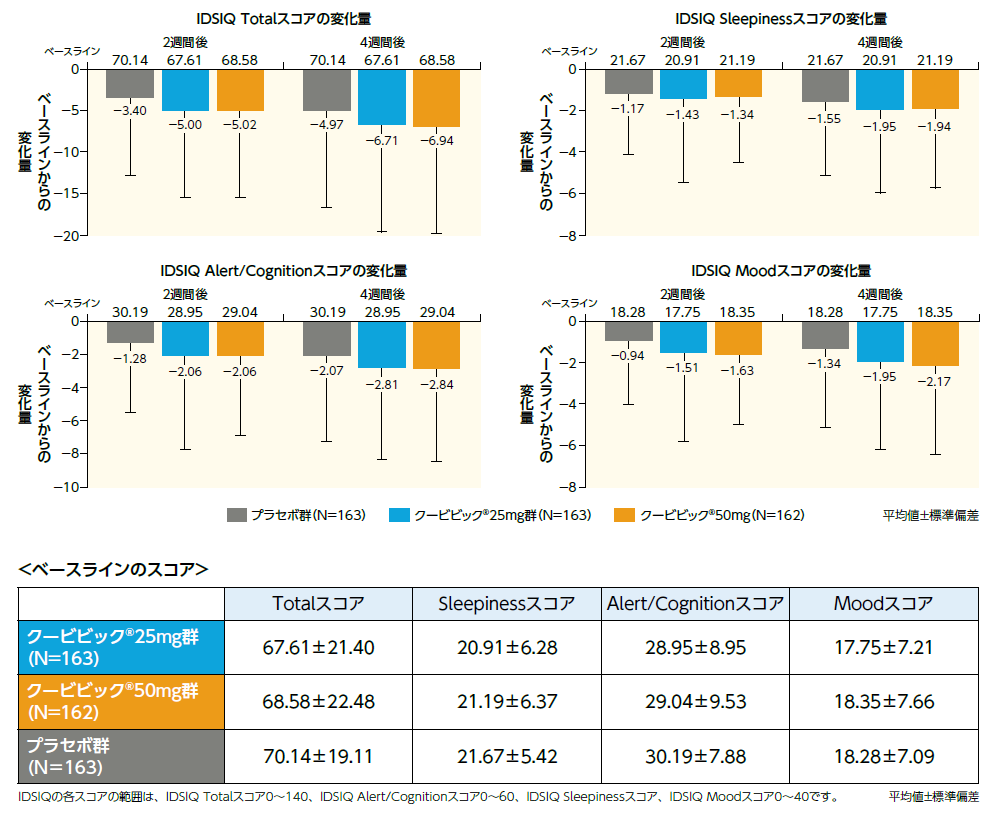
IDSIQ(探索的評価項目)
安全性
二重盲検期において、有害事象はクービビック25mg群で17.8%(29/163例)、50mg群で22.2%(36/162例)、プラセボ群で22.6%(37/164例)に、副作用はそれぞれ7.4%(12/163例)、11.1%(18/162例)及び8.5%(14/164例)に発現しました。主な副作用は傾眠[25mg群3.7%(6/163例)、50mg群6.8%(11/162例)、プラセボ群1.8%(3/164例)]でした。
二重盲検期に投与中止に至った副作用は、25mg群で1例(0.6%)に発疹、50mg群で1例(0.6%)に浮動性めまいが報告されました。
本試験において、重篤な副作用、死亡に至った副作用は報告されませんでした。
MedDRA version 25.0
国内第Ⅲ相長期投与試験(ID-078A305試験)
試験概要
- 目的
-
日本人不眠症患者におけるクービビック長期投与の安全性を評価する。
- 試験デザイン
-
多施設共同、ランダム化、非盲検試験
- 対象
-
不眠症患者154例
<主な登録基準>
・18歳以上の男女
・DSM-5に基づき不眠障害と診断された患者
・不眠重症度指数(ISI)スコアが15以上の患者
・普段の就床時刻が21:30~00:30、就床時間が6時間から9時間の患者
・ランダム化(来院2回目)の前7日間に自宅で記入した睡眠日誌において、3夜以上で以下のすべてに該当する患者
(a)主観的睡眠潜時(sLSO)30分以上 (b)主観的中途覚醒時間(sWASO)30分以上
(c)主観的総睡眠時間(sTST)6.5時間以下
・睡眠関連呼吸障害(慢性閉塞性肺疾患、睡眠時無呼吸を含む)の既往歴がなく、合併のない患者
・周期性四肢運動障害、レストレスレッグス症候群、概日リズム睡眠障害、レム睡眠行動障害、ナルコレプシーをいずれも合併していない患者
・スクリーニング前12週以内に精神疾患のエピソードがなかった患者 - 方法
-
スクリーニング期(1~4週間)の後、クービビック25mg群又は50mg群に1:2の比でランダムに割付け、非盲検下で治験薬を1日1回就寝前に52週間経口投与した。割付けは、高齢者(65歳以上)、非高齢者を層別因子とした。
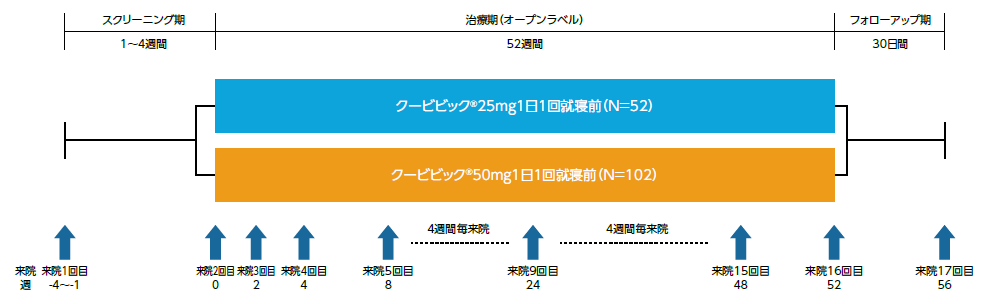
- 評価項目
-
安全性評価項目(主要評価項目):
有害事象、バイタルサイン、臨床検査、心電図、C-SSRS、翌日の持ち越し効果(睡眠日誌のVAS「今朝のあなたの気分について」)、日中の眠気(JESS)等
有効性評価項目(副次評価項目):
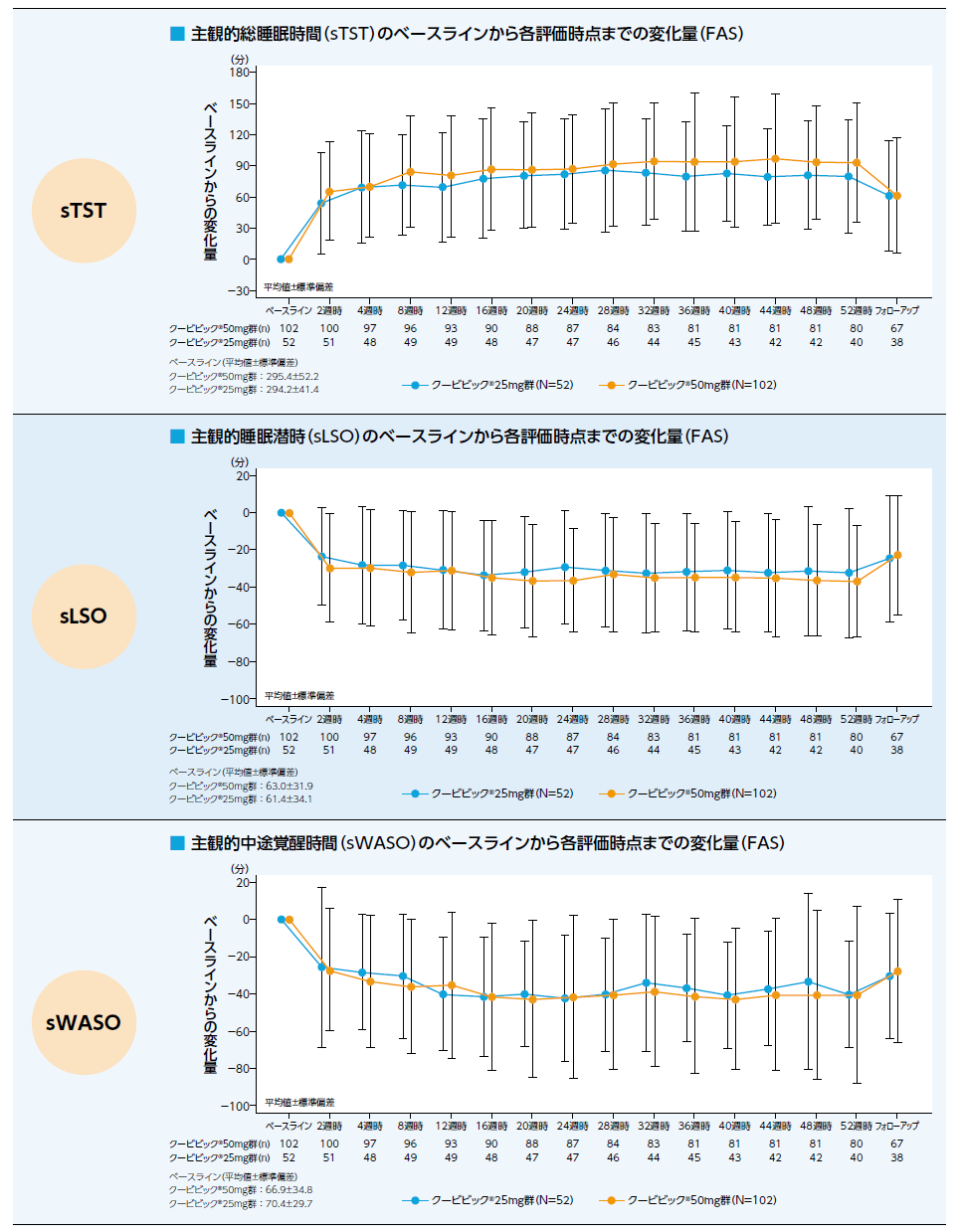
・以下の項目の各評価時点*1におけるベースライン*2からの変化量
主観的総睡眠時間(sTST)、主観的睡眠潜時(sLSO)、主観的中途覚醒時間(sWASO) 等
探索的評価項目:
・IDSIQの各スコアの各評価時点*3におけるベースライン*2からの変化量
*1:評価日及びその直前の6日間における平均値
*2:ランダム化(来院2回目)の前7日間に記入した睡眠日誌/不眠症の日中の症状及び影響に関する質問票(IDSIQ)に基づく平均値
*3:評価日直前の7日間の平均値 - 解析計画
-
解析対象集団:
有効性の解析対象集団は治験薬を1回以上投与されたすべての患者(FAS)、安全性の解析対象集団は治験薬を1回以上投与されたすべての患者とした。
有効性評価項目:
各評価項目の測定値及びベースラインからの変化量について、要約統計量を算出した。評価項目の各評価時点の週平均は、IDSIQ及びVASは評価日直前の7日間の平均値、それ以外は評価日及びその直前の6日間における平均値とし、1週間のデータが3日未満であった場合、その週平均は欠測とした。
サブグループ解析:
主観的総睡眠時間(sTST)及び主観的睡眠潜時(sLSO)について、併存疾患(あり、なし)、併存疾患のクラス(精神疾患、精神疾患以外)及び年齢区分(65歳未満、65歳以上)別にサブグループ解析を実施することを事前に計画した。
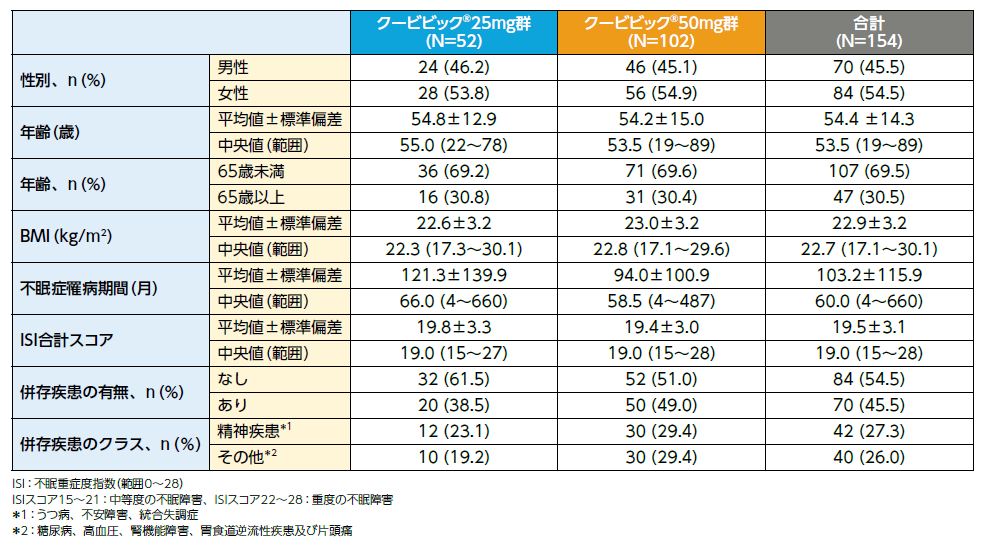
患者背景(FAS)
安全性(主要評価項目)
有害事象はクービビック25mg群57.7%(30/52例)、50mg群73.5%(75/102例)に、副作用は25mg群19.2%(10/52例)、50mg群24.5%(25/102例)に発現しました。
主な有害事象(いずれかの投与群で発現頻度5%以上)は、発熱[25mg群17.3%(9/52例)、50mg群15.7%(16/102例)]、傾眠[25mg群13.5%(7/52例)、50mg群14.7%(15/102例)]、頭痛[25mg群11.5%(6/52例)、50mg群14.7%(15/102例)]、倦怠感[25mg群0%、50mg群8.8%(9/102例)]、上咽頭炎[25mg群5.8%(3/52例)、50mg群2.0%(2/102例)]、主な副作用(いずれかの投与群で発現頻度5%以上)は、傾眠[25mg群11.5%(6/52例)、50mg群13.7%(14/102例)]、倦怠感[25mg群0%、50mg群5.9%(6/102例)]でした。
重篤な有害事象は、25mg群5.8%(3/52例)、50mg群5.9%(6/102例)に認められ、内訳は25mg群で変形性関節症、ギラン・バレー症候群、特発性肺線維症が各1例(1.9%)、50mg群でデュプイトラン拘縮、不安定狭心症、胆管炎、COVID-19、硬膜下血腫、大動脈解離が各1例(1.0%)でした。いずれも治験薬との関連性は否定されました。
投与中止に至った有害事象は、25mg群11.5%(6/52例)、50mg群6.9%(7/102例)に認められ、内訳は25mg群で傾眠2例(3.8%)、ギラン・バレー症候群、COVID-19、妊娠時曝露及び特発性肺線維症が各1例(1.9%)、50mg群で傾眠、倦怠感、頭痛、頚髄神経根障害、COVID-19、コロナウイルス感染、胆管炎及び関節周囲炎が各1例(1.0%)でした。治験薬との関連ありと判定された事象は、25mg群で傾眠2例(3.8%)、50mg群で傾眠、倦怠感及び頭痛が各1例(1.0%)でした。
本試験においていずれの群でも死亡は認められませんでした。
MedDRA version 25.0
有効性のまとめ:副次評価項目
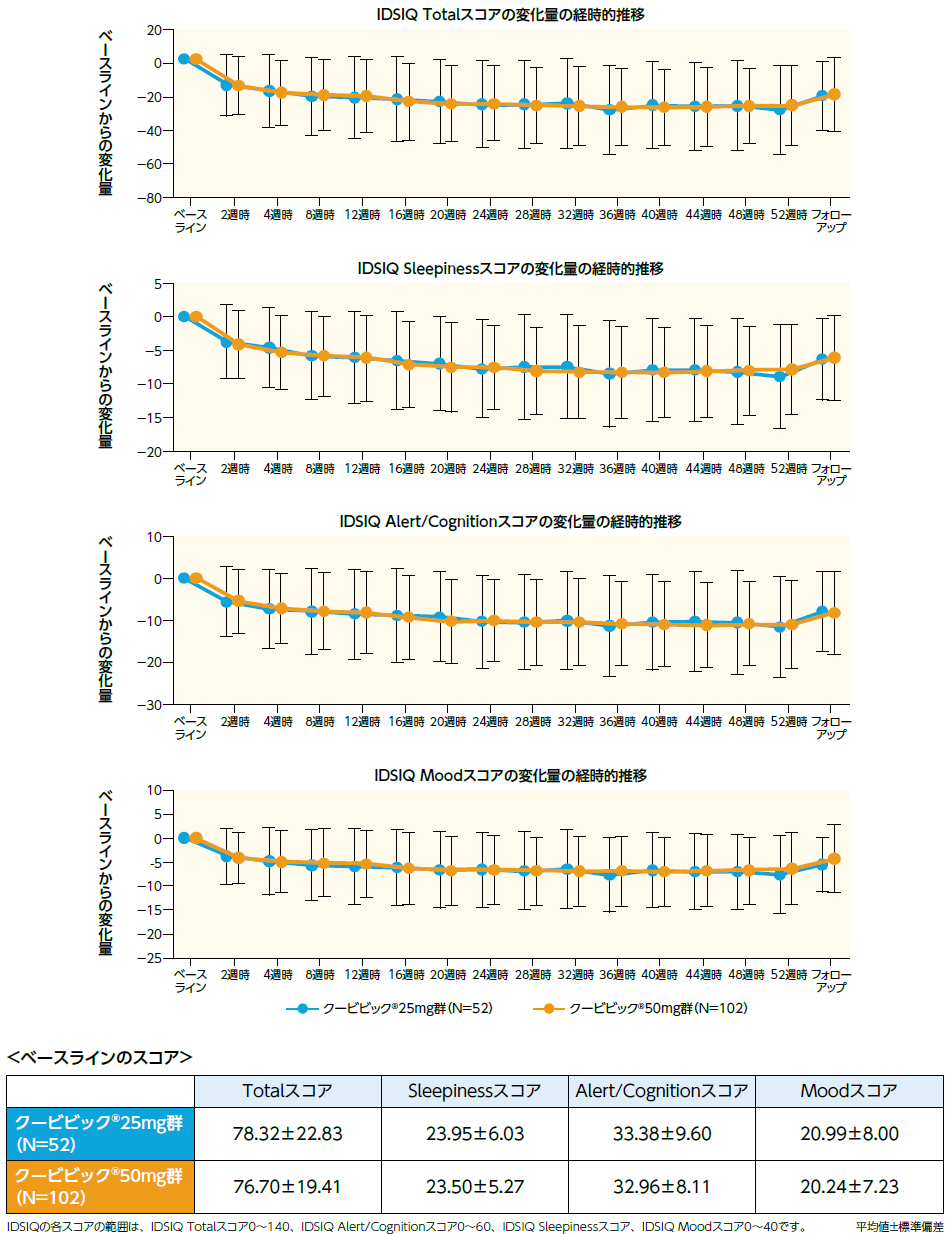
IDSIQ(探索的評価項目)
海外第Ⅲ相試験(ID-078A301):海外データ
試験概要
- 目的
-
不眠障害患者を対象としたクービビックの有効性及び安全性を検討する。
- 試験デザイン
-
多施設共同、二重盲検、ランダム化、プラセボ対照、並行群間、睡眠ポリグラフ(PSG)試験
- 対象
-
不眠障害を有する成人及び高齢患者930例
<主な登録基準>
・18歳以上、BMIが18.5kg/m2以上40.0kg/m2以下の男女
・DSM-5に基づき不眠障害と診断された患者
・来院1回目の不眠重症度質問票(ISI)スコアが15以上の患者
・来院2回目から3回目の期間7日間で記入した睡眠日誌において、3夜以上で以下のすべてに該当する患者
(a)主観的睡眠潜時(sLSO)30分以上 (b)主観的中途覚醒時間(sWASO)30分以上
(c)主観的総睡眠時間(sTST)6.5時間以下
・来院2回目から3回目の期間7日間で記入した睡眠日誌において、普段の就床時刻が21:30~00:30、就床時間が6時間から9時間と報告した患者
・来院3回目に2夜のPSGにより測定した睡眠パラメータが以下の基準をすべて満たす患者
(a)持続入眠潜時(LPS)の平均値が20分以上(2夜のいずれでも15分未満ではない)
(b)中途覚醒時間(WASO)の平均値が30分以上(2夜のいずれでも20分未満ではない)
(c)総睡眠時間(TST)の平均値が420分未満
・慢性閉塞性肺疾患及び睡眠時無呼吸などの睡眠関連呼吸障害の既往歴がなく、合併のない患者
・急性又は不安定な精神状態(不安障害、大うつ病、双極性障害、統合失調症、強迫性障害、又はうつ病を含むが、これらに限定されない)と診断されていない患者、大うつ病の既往歴を有するが、無症状で、同意取得時に治療不要であった患者 - 方法
-
被験者は、スクリーニング期(7~18日間)と、これに続く単盲検のプラセボrun-in期(13~24日間)の後、クービビック25mg群、50mg群又はプラセボ群に、それぞれ1:1:1の比でランダムに割付けられた。ランダムに割付け後、二重盲検下にて治験薬を1日1回就寝前に12週間経口投与し、その後7日間を単盲検にてプラセボ投与するプラセボrun-out期とした。
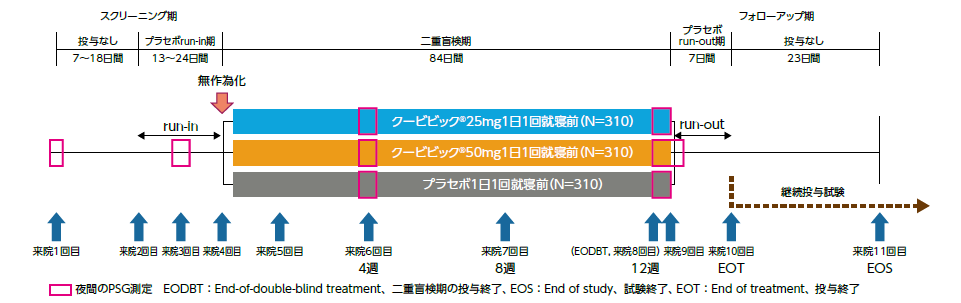
- 評価項目
-
主要評価項目(有効性):検証的解析項目
・中途覚醒時間(WASO)の4週時注1) 及び12週時注1) におけるベースライン注2) からの変化量
・持続入眠潜時(LPS)の4週時注1) 及び12週時注1) におけるベースライン注2) からの変化量
副次評価項目(有効性):
・主観的総睡眠時間(sTST)の4週時注3) 及び12週時注3) におけるベースライン注4) からの変化量
・IDSIQ Sleepinessスコアの4週時注3) 及び12週時注3) におけるベースライン注4) からの変化量
探索的評価項目:
・IDSIQの各スコア(Sleepinessスコア以外)の4週時及び12週時注3) におけるベースライン注4) からの変化量
注1)4週時、12週時:来院6回目、8回目それぞれの2夜PSG測定での平均値
注2)ベースライン:来院3回目の2夜PSG測定での平均値
注3)4週時、12週時:来院6回目、8回目それぞれの最初のPSG測定の直前7日間に被験者が自宅で実施したSDQ/IDSIQの記入データにおける平均値
注4)ベースライン:来院3回目の最初のPSG測定の直前7日間に被験者が自宅で実施したスクリーニングSDQ/IDSIQの記入データにおける平均値
安全性評価項目:
・二重盲検期終了(中止)後30日又は継続投与試験に移行するまでに治療下で発現した有害事象(TEAE)
・二重盲検期終了(中止)後30日又は継続投与試験に移行するまでに発現した重篤な有害事象
・二重盲検期の早期治験薬投与中止に至った有害事象 等
- 解析計画
-
解析対象集団:
有効性の解析対象集団は、ランダム化され少なくとも1回治験薬を投与されたすべての患者(FAS)、安全性の解析対象集団は治験薬を1回以上投与されたすべての患者とした。
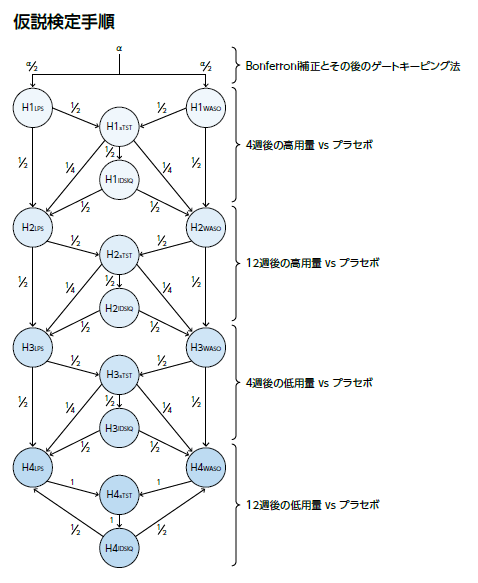
主要評価項目(WASO及びLPS)及び副次評価項目(sTST及びIDSIQ Sleepinessスコア)それぞれについて、2用量に対する計16の帰無仮説検定を実施した。両側有意水準5%で試験全体の第1種過誤を制御するため、 Bonferroni法に基づくゲートキーピング法を用い、主要及び副次評価項目のそれぞれ2つの項目を両側 5%の半分の有意水準で、それぞれ4週時の高用量群とプラセボ群の比較、12週時の高用量群とプラセボ群の比較、4週時の低用量群とプラセボ群の比較、12週時の低用量群とプラセボ群の比較の順で検定を行った。
評価項目のベースラインからの変化量は、各応答変数(WASO、LPS、sTST、又はIDSIQ sleepinessスコア)のベースライン値、年齢区分(65歳未満、65歳以上)、投与群(高用量群、低用量群、プラセボ)、評価時点(4週時、12週時)、投与群と評価時点の交互作用項及びベースライン値と評価時点の交互作用項を適用した線形混合効果モデルを用いて解析し、対象の群間差を検定した。

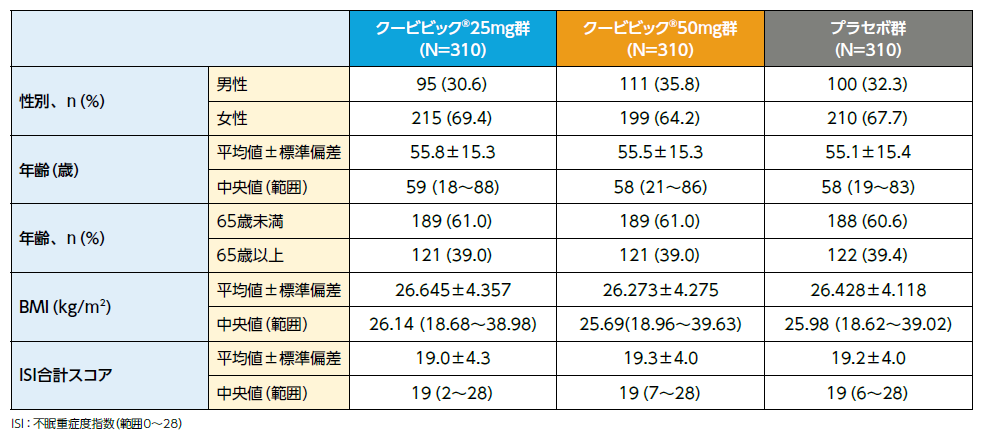
患者背景(FAS)
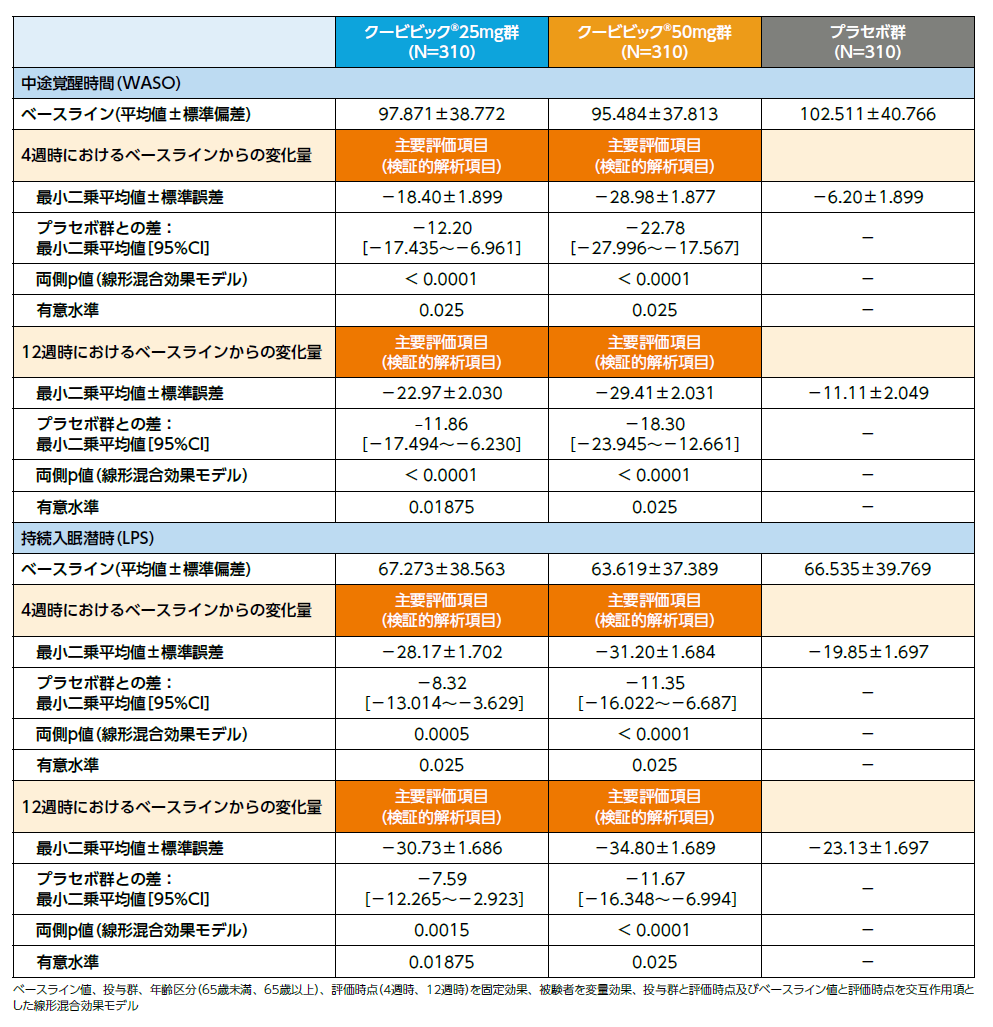
有効性のまとめ:主要評価項目(検証的解析結果)、副次評価項目
クービビック25mg群、クービビック50mg群の4週時、12週時において、プラセボ群に対する中途覚醒時間(WASO)の優越性が検証されました(いずれもp<0.0001、線形混合効果モデル)。
クービビック25mg群、クービビック50mg群の4週時、12週時において、プラセボ群に対する持続入眠潜時(LPS)の優越性が検証されました(25mg群4週時:p=0.0005、25mg群12週時:p=0.0015、50mg群4週時、12週時ともにp<0.0001、線形混合効果モデル)。
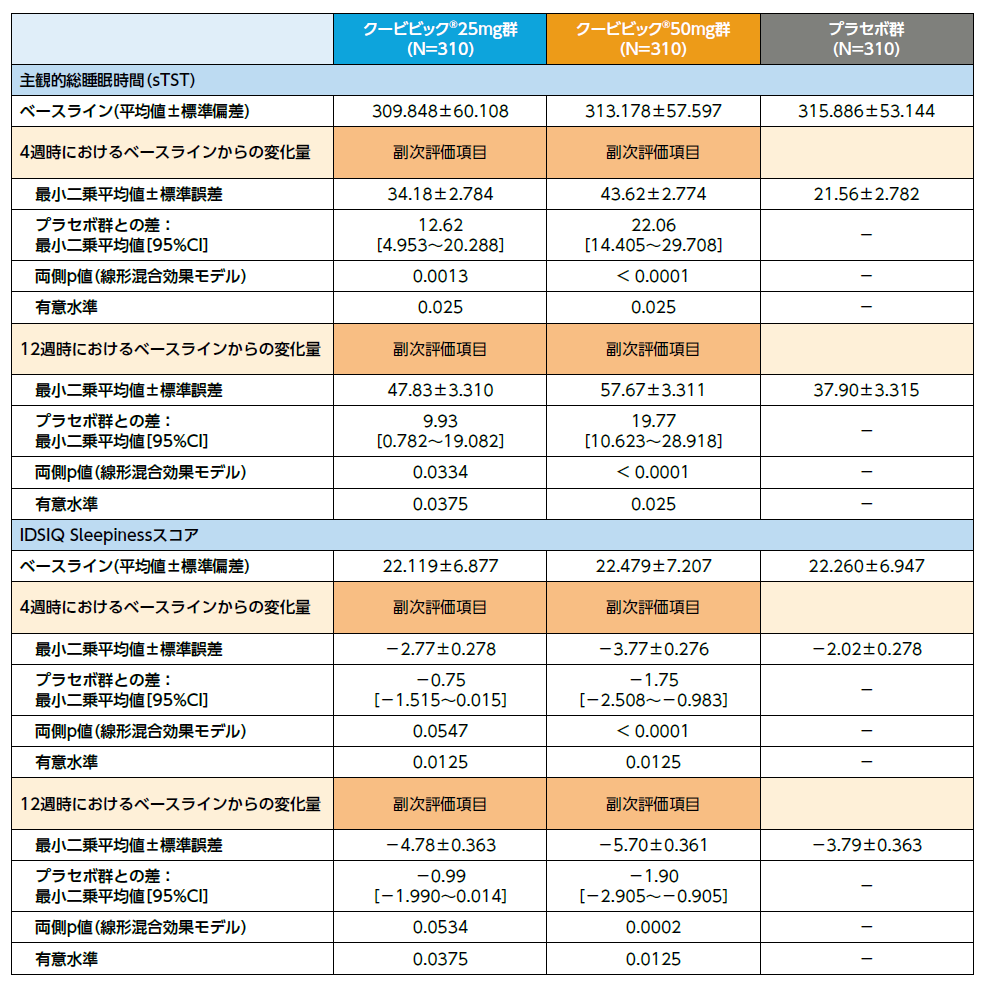
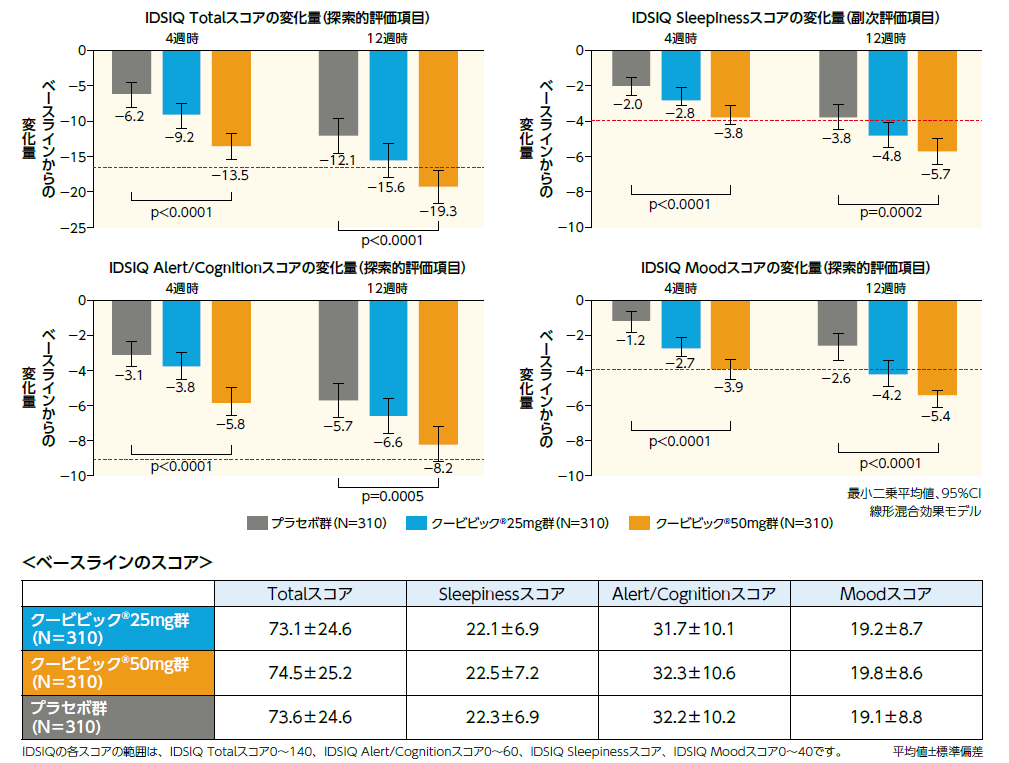
IDSIQ(副次評価項目、探索的評価項目)
副次評価項目であるIDSIQ Sleepinessスコアの4週時及び12週時におけるベースラインからの変化量のプラセボ群との比較において、クービビック50mg群で有意な低下が認められました。
探索的評価項目であるTotalスコア、Alert/Cognitionスコア、Moodスコアの4週時及び12週時におけるベースラインからの変化量のプラセボ群との比較において、クービビック50mg群で有意な低下が認められました(いずれも多重性を考慮していない名目上の有意差あり)。
意味のある変化量(赤破線)8)
●Totalスコア 17点 ●Sleepinessスコア 4点 ●Alert/Cognitionスコア 9点 ● Moodスコア 4点
意味のある変化量を算出するために、海外の承認時評価試験とされた海外第Ⅲ相試験(ID-078A301)9) のデータを用いました。被験者は、二重盲検治療期間を通じて、毎日夕方にIDSIQの記録を行い、スコアは週平均として計算されました(各IDSIQ項目の評価スケールについては、「日中の精神・身体機能を評価する指標 ~IDSIQとは~」をご参照ください)。
患者による疾患重症度の全般評価(PGA-S)、患者による重症度の全般的印象(PGI-S)、患者による変化の全般的印象(PGI-C)及びISI合計スコアは、治療前後の患者の有意な変化を推定するための潜在的なアンカーとして評価され、アンカーのスコア変化/評価とIDSIQの関係に関するスピアマン相関係数(4週時で0.36~0.44、12週時で0.45~0.57)は、すべて事前に規定された閾値0.30を上回りました。また、PGA-S、PGI-Sでは1~2点の減少、PGI-Cでは「少し改善した」「中程度に改善した」、ISIは6点の減少が、「臨床的に関連するスコアの変化」とされており10,11) 、その閾値に達した患者のIDSIQスコアに基づき、意味のある変化量が算出されました。
本解析については以下のlimitationが存在します8) 。
●対象患者は中等度から重度の不眠症患者であり、日常臨床で治療を受けている多くの患者を代表するものではない可能性がある。
●対象患者は主に白人であったため、人種的・民族的多様性を十分に反映していない可能性がある。
●対象となった臨床試験では、不眠症による日中の症状のベースラインの基準は設定していなかったため、一般的な不眠症患者の日中の症状を反映していない可能性がある。
●試験期間(12週)だけでは、治療による患者の変化を正確に反映していない可能性がある。
安全性
二重盲検期において、有害事象はク-ビビック25mg群で37.7%(117/310例)、50mg群で37.7%(116/308例)、プラセボ群で34.0%(105/309例)に、副作用は25mg群で12.9%(40/310例)、50mg群で12.3%(38/308例)、プラセボ群9.4%(29/309例)に発現しました。主な副作用は傾眠[25mg群2.6%(8/310例)、50mg群1.6%(5/308例)、プラセボ群1.6%(5/309例)]でした。
重篤な副作用は、本試験においてクービビック群では認められず、プラセボ群で失神が1例に認められました。
二重盲検期に投与中止に至った副作用は25mg群で1.9%(6/310例)、50mg群で0.6%(2/308例)、プラセボ群で1.3%(4/309例)に認められました。その内訳は、25mg群で浮動性めまい2例(0.6%)、抑うつ気分、鎮静合併症、睡眠の質低下、睡眠時麻痺が各1例(0.3%)、50mg群で上室性期外収縮、腎機能障害が各1例(0.3%)、プラセボ群で失神、耳鳴、不眠症、鎮静合併症が各1例(0.3%)でした。
本試験において、死亡に至った副作用は報告されませんでした。
MedDRA version 22.1
不眠症治療のゴールは、不眠症状が十分に消褪しているとともに、QOL障害が改善していること12)
監修
秋田大学大学院医学系研究科 医学専攻 病態制御医学系 精神科学講座
三島 和夫 先生*
*資材ご監修時のお肩書で掲載させていただいております。

ICSD-3-TR(睡眠障害国際分類 第3版 改訂版)やICD‑11(国際疾病分類第11版)では、不眠障害(不眠症)とは夜間の不眠症状 sleep disturbanceに加えて、不眠に起因する日中機能の障害 daytime impairment(認知・社会機能障害、QOL障害)が生じる疾患とされています。逆に、不眠症状があっても心身の不調が無く元気に生活している人も少なからずおられます。実際、私自身も不眠診療を行っていると、不眠症状の改善だけでは不十分で、日中機能障害が回復してはじめて患者さんに笑顔が戻ることを実感しています。眠れるようになることも大事ですが、そのことで朝に気持ちよく目覚め、疲労回復感があり、日中に心身ともにスッキリとした状態で生活できることが治療のゴールであると考えます。そのため、以前の不眠症治療薬の臨床試験においては、夜間の不眠症状の改善を中心に薬効を評価することが一般的でしたが、「睡眠薬の臨床評価方法に関するガイドライン」13) では日中の機能障害の評価を行う必要があることが記載されています。
クービビック錠の臨床試験では日中機能の評価が行われています。そのためにFDAの患者報告アウトカム(Patient‑Reported Outcome:PRO)ガイダンスに従い新たに日中機能の評価スケールであるIDSIQが開発されました。不眠症治療薬によって夜間の不眠症状の改善だけでなく、日中機能を積極的に評価するための新たな取り組みで、高く評価されるべきでしょう。本資材では、IDSIQを用いて実施された国内の2試験及び海外の1試験を紹介しています。いずれの臨床試験でも不眠重症度指数(ISI)が15以上の患者が登録条件となっていましたが、国内第Ⅲ相試験(ID‑078A304)に登録された患者の平均ISIは17であり、ほかの2試験よりも軽症の患者が対象となっていました。同時にベースライン時のIDSIQスコアも各臨床試験間で違いがみられました。他の要因も含めて総合的に試験結果を読み解く必要があります。
今後の不眠診療にあたっては、昼と夜の両方の症状(困りごと)に着目して診断し、薬効を評価し、治療計画を組み立てていただきたいと思います。その際に患者さんの日中機能について考慮された患者日誌などもご検討ください。
References
1) 三島 和夫, 精神保健研究 2016; 62: 81‑89.
2) American Academy of Sleep Medicine, 日本睡眠学会 診断分類委員会 監訳, 睡眠障害国際分類 第3版 改訂版, ライフサイエンス出版, 2025, pp1, 3, 19
3) 睡眠障害の診断・治療ガイドライン研究会 内山 真 編, 睡眠障害の対応と治療ガイドライン 第3版, じほう, 2019, p161
4) 伊藤 結生 ほか, 精神医学 2022; 64(3): 333‑340.
5) Buysse DJ, et al.: Sleep Med. 2007; 8(3): 198‑208.
6) Hudgens S, et al.: Patient. 2021; 14(2): 249‑268.(本研究はイドルシア社の資金により行われた。本論文の著者のうち1名はイドルシア社の社員である。)
7) 社内資料:主な他の有効性評価(承認時評価資料)
8) Phillips-Beyer A, et al.: Pharmaceut Med. 2023; 37(4): 291-303.(本研究はイドルシア社の支援により実施された。)
9) 社内資料:不眠症患者を対象とした海外第Ⅲ相試験[ID-078A301試験](承認時評価資料)
10) Yang M, et al.: Curr Med Res Opin. 2009; 25(10): 2487-94.
11) Omachi TA.: Arthritis Care Res (Hoboken). 2011; 63 Suppl 11(0 11): S287-96.
12) 厚生労働科学研究班・日本睡眠学会ワーキンググループ作成「睡眠薬の適正な使用と休薬のための診療ガイドライン」
13) 睡眠薬の臨床評価方法に関するガイドライン(平成23年12月13日薬食審査発1213第1号、厚生労働省医薬食品局審査管理課長通知別添) https://www.pmda.go.jp/files/000208186.pdf(2025年10月閲覧)

