クービビックの臨床成績
クービビックの特性
1.クービビックは、オレキシン受容体タイプ1(OX1R)及びオレキシン受容体タイプ2(OX2R)の両受容体に作用するデュアルオレキシン受容体拮抗薬であり、オレキシン神経ペプチドによる OX1R及びOX2Rの活性化をともに阻害します。
3.日本人不眠症患者において、クービビック1日1回就寝前投与により主観的及び客観的睡眠パラメータの改善が認められました。主観的総睡眠時間(sTST)の延長及び主観的睡眠潜時(sLSO)の短縮が認められました。
国内第Ⅲ相試験[ID-078A304試験](検証的試験):
・クービビック50mg群において、主観的総睡眠時間(sTST)及び主観的睡眠潜時(sLSO)のいずれも4週時におけるベースラインからの変化量について、プラセボ群と比べ有意な改善が認められました(いずれもp<0.001、線形混合効果モデル)(主要評価項目、検証的解析結果)
・クービビック25mg群において、sTST及びsLSOのいずれも4週時におけるベースラインからの変化量について、プラセボ群と比べ有意な改善が認められました(それぞれp=0.042、p=0.006、線形混合効果モデル)(副次評価項目)
国内第Ⅱ相用量反応試験[ID-078A206試験](検証的試験)1) :
・睡眠ポリグラフ(PSG)で測定した中途覚醒時間(WASO)及び持続入眠潜時(LPS)のクービビック投与1日目及び2日目(2夜連続PSGの平均値)におけるベースラインからの変化量は、有意な用量反応性が認められ(WASO:主要評価項目、p<0.0001、検証的解析結果、 Emaxモデル/ LPS:副次評価項目、p<0.0001、名目上のp値、線形モデル)、クービビック50mg群及び25mg群ともにプラセボ群と比べ有意に改善しました(WASOの補助的解析、すべてp<0.05、名目上のp値、線形混合効果モデル)
電子化された添付文書の副作用及び臨床成績の安全性の結果をご参照ください。
- 4. 効能又は効果
-
不眠症
- 6. 用法及び用量
-
通常、成人にはダリドレキサントとして1日1回50mgを就寝直前に経口投与する。なお、患者の状態に応じて1日1回25mgを投与することができる。
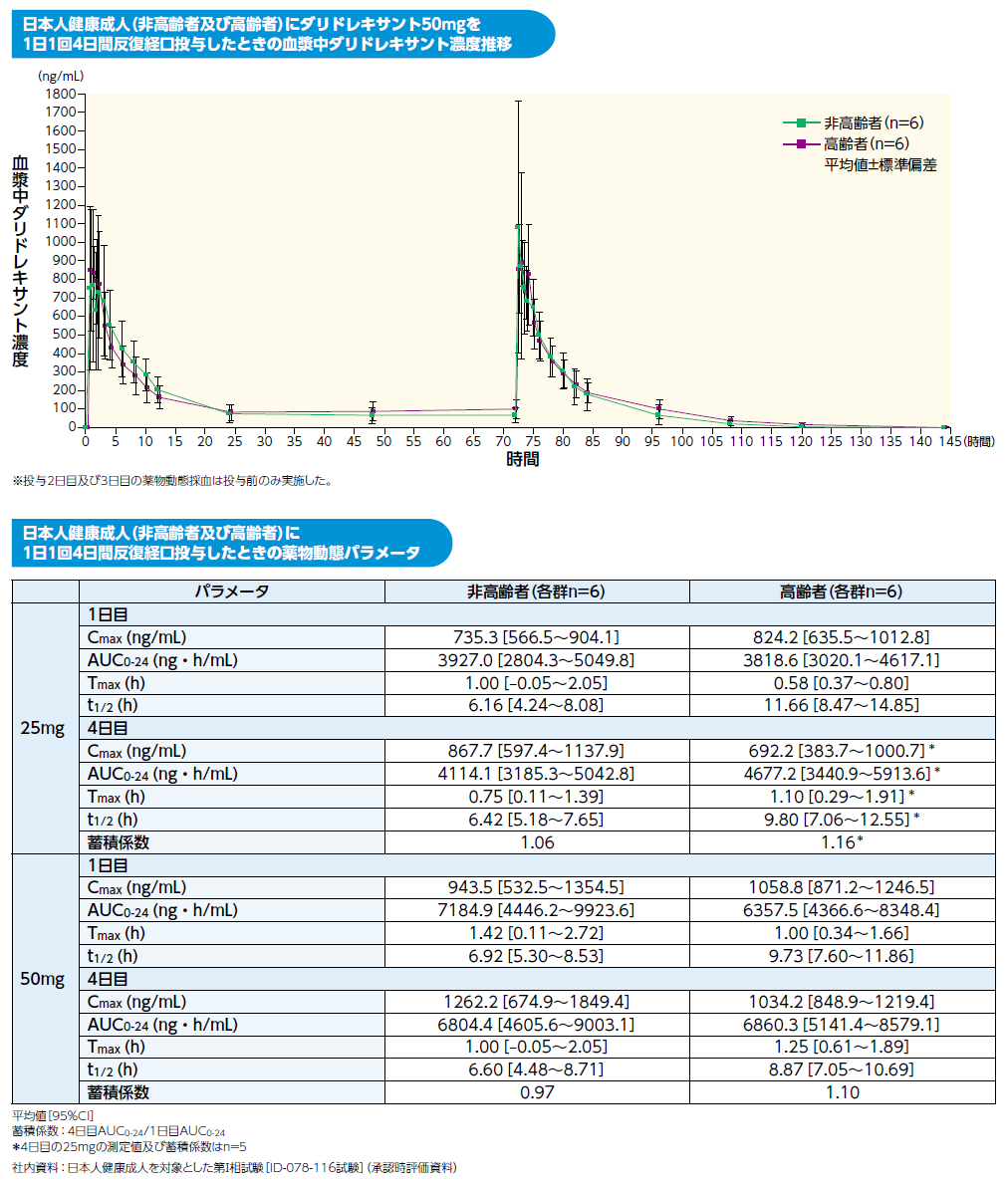
クービビックの薬物動態
国内第Ⅲ相試験(ID-078A304試験)
社内資料:日本人不眠症患者を対象とした国内第Ⅲ相試験[ID-078A304試験](承認時評価資料)
クービビック錠審査報告書
Uchimura N, et al.: Sleep Med. 2024; 122: 27-34.
( 本研究はNxera Pharma Japanの資金により行われた。著者はNxera Pharma Japanよりコンサルタント料等を受領している。
著者はNxera Pharma Japan社員である。)( 本研究はMochida Pharmaceutical Co., Ltd.の資金により行われた。)
①試験概要
- 目的
-
日本人不眠症患者におけるクービビックの有効性及び安全性を評価する。
- 試験デザイン
-
多施設共同、無作為化、二重盲検、プラセボ対照、並行群間比較試験
- 対象
-
不眠症患者490例
<主な登録基準>
・18歳以上、BMIが30.0kg/m2未満の男女
・DSM-5に基づき不眠障害と診断された患者
・ 初回スクリーニング(来院1回目)前の12週以上にわたって週3夜以上、自己報告による以下のすべての症状がある患者
(a)主観的睡眠潜時(sLSO)30分以上 (b)主観的中途覚醒時間(sWASO)30分以上 (c)主観的総睡眠時間(sTST)6.5時間以下
・来院1回目の不眠重症度指数(ISI)スコアが15以上の患者
・来院1回目で普段の就床時刻(中央値)が20:30~00:30、就床時間(中央値)が6時間から9時間と報告した患者
・プラセボrun-in期(来院2回目)の直前7日間記入した睡眠日誌において、3夜以上で以下のすべてに該当する患者
(a)主観的睡眠潜時(sLSO)30分以上 (b)主観的中途覚醒時間(sWASO)30分以上 (c)主観的総睡眠時間(sTST)6.5時間以下
・ 来院2回目の直前7日間記入した睡眠日誌において、普段の就床時刻(中央値)が20:30~00:30、就床時間(中央値)が6時間から9時間と報告した患者
・ 無作為化(来院3回目)の直前7日間のうち6日以上プラセボを服用し、睡眠日誌においてプラセボ服用日のうち3夜以上で以下のすべてに該当する患者
(a)主観的睡眠潜時(sLSO)30分以上 (b)主観的中途覚醒時間(sWASO)30分以上 (c)主観的総睡眠時間(sTST)6.5時間以下
・ 来院3回目の直前7日間記入した睡眠日誌において、普段の就床時刻(中央値)が20:30~00:30、就床時間(中央値)が6時間から9時間と報告した患者
・ 睡眠関連呼吸障害(慢性閉塞性肺疾患等)、睡眠時無呼吸症候群の既往歴がなく、合併していない患者
・ 周期性四肢運動障害、レストレスレッグス症候群、概日リズム睡眠障害、レム睡眠行動障害、ナルコレプシーをいずれも合併していない患者、精神疾患(不安障害、大うつ病、双極性障害、統合失調症、強迫性障害等)を有していない患者
- 方法
-
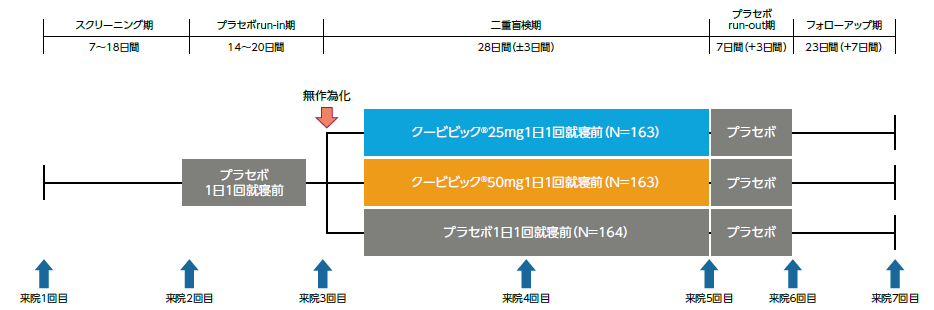
本試験はスクリーニング期(7~18日間)、単盲検のプラセボrun-in期(14~20日間)、二重盲検期(28日間)、プラセボrun-out期(7日間)及びフォローアップ期(23日間)で構成された。
プラセボrun-in期の後、クービビック25mg群、50mg群又はプラセボ群に1:1:1の比で無作為に割付け、二重盲検下で1日1回就寝前に4週間経口投与した。その後、単盲検にてプラセボを1日1回就寝前に7日間経口投与した。
- 評価項目
-
主要評価項目(有効性):検証的解析項目
・4週時における主観的総睡眠時間(sTST)ならびに主観的睡眠潜時(sLSO)のベースラインからの変化量のクービビック50mg群とプラセボ群の比較
副次評価項目(有効性):
・4週時における主観的総睡眠時間(sTST)ならびに主観的睡眠潜時(sLSO)のベースラインからの変化量のクービビック25mg群とプラセボ群の比較
探索的評価項目:
・ 2週時における主観的総睡眠時間(sTST)のベースラインからの変化量のクービビック25mg群、50mg群とプラセボ群の比較
・ 2週時における主観的睡眠潜時(sLSO)のベースラインからの変化量のクービビック25mg群、50mg群とプラセボ群の比較
・ 4週時における主観的中途覚醒時間(sWASO)のベースラインからの変化量
・ 4週時における主観的睡眠効率(100×sTST/眠ろうとし始めた時から最後に目が覚めた時までの時間)のベースラインからの変化量
・ 4週時における中途覚醒回数のベースラインからの変化量 等
安全性評価項目:
有害事象、バイタルサイン、臨床検査、心電図、C-SSRS、反跳性不眠、退薬症候[ベンゾジアゼピンによる離脱症状に関する質問票(BWSQ)]、日中の眠気[エプワース眠気尺度日本語版(JESS)]、翌日の持ち越し効果[WAIS-IV知能検査(符号)] 等
- 解析計画
-
解析対象集団:
有効性の解析対象集団は、無作為化され少なくとも1回治験薬を投与されたすべての患者(FAS)、安全性の解析対象集団は治験薬を1回以上投与されたすべての患者とした。
主要評価項目(2週時は探索的評価項目):
主観的総睡眠時間(sTST)及び主観的睡眠潜時(sLSO)のベースラインからの変化量について、それぞれのベースライン値、投与群、年齢区分(65歳未満、65歳以上)、評価時点(2週時、4週時)を固定効果、被験者を変量効果、投与群と評価時点及びベースライン値と評価時点を交互作用項とした線形混合効果モデルを用いて解析した。線形混合効果モデルは、無構造共分散行列を基本として測定間の相関をモデル化し、対比を用いてプラセボ群とクービビック50mg群を比較した。
検定の多重性による第1種の過誤の増大を防ぐため、階層的閉手順により4週時における主観的総睡眠時間(sTST)についてプラセボ群とクービビック50mg群との比較を行い、両側有意水準0.05で有意差が認められた場合、4週時における主観的睡眠潜時(sLSO)についてプラセボ群とクービビック50mg群を比較した。
なお、主観的総睡眠時間(sTST)及び主観的睡眠潜時(sLSO)とも、評価時点(2週時、4週時)の週平均は、評価日及びその直前の6日間(少なくとも3日間)における平均値とし、1週間のデータが3日未満であった場合、その週平均は欠測とし、主要解析においては欠測値の補完は行わず、線形混合効果モデルを適用した。
副次評価項目:
主要評価項目である4週時における主観的睡眠潜時(sLSO)の帰無仮説が棄却された場合、検定の多重性による第1種の過誤の増大を防ぐため、階層的閉手順により4週時における主観的総睡眠時間(sTST)についてプラセボ群とクービビック25mg群との比較を行い、両側有意水準0.05で有意差が認められた場合、4週時における主観的睡眠潜時(sLSO)についてプラセボ群とクービビック25mg群を比較した。主要評価項目の解析で用いるモデルを用い、対比によりプラセボ群とクービビック25mg群を比較した。
探索的評価項目:
連続量については要約統計量(例数、平均、標準偏差、中央値、最小値、最大値)を算出し、分類データについては例数とその割合、必要に応じて発現割合の95%CIを算出した。
サブグループ解析:
主要評価項目及び副次評価項目について、年齢区分(65歳未満、65歳以上)、性別(男性、女性)、併存疾患(あり、なし)によりサブグループ解析を実施することを事前に計画した。
事後解析:
本邦における承認審査の過程において、PMDAの指示により2週時及び4週時における主観的中途覚醒時間(sWASO)のベースラインからの変化量は最小二乗平均値を用いて再解析を行った。主要評価項目の解析で用いるモデルを用い、対比によりプラセボ群とクービビック25mg群、50mg群を比較した。
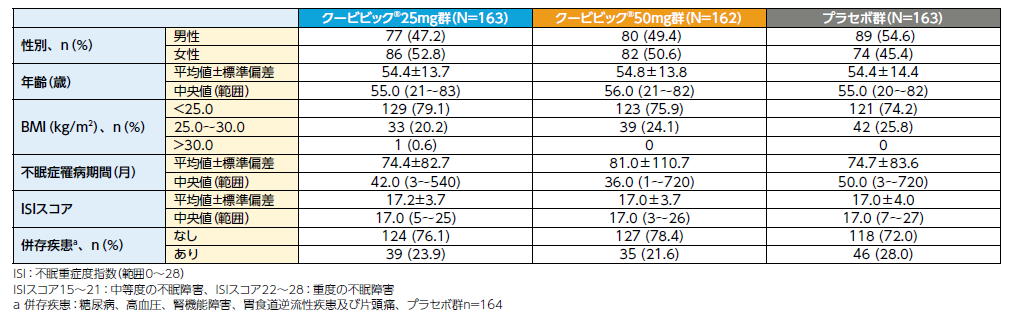
②患者背景(FAS)
③有効性
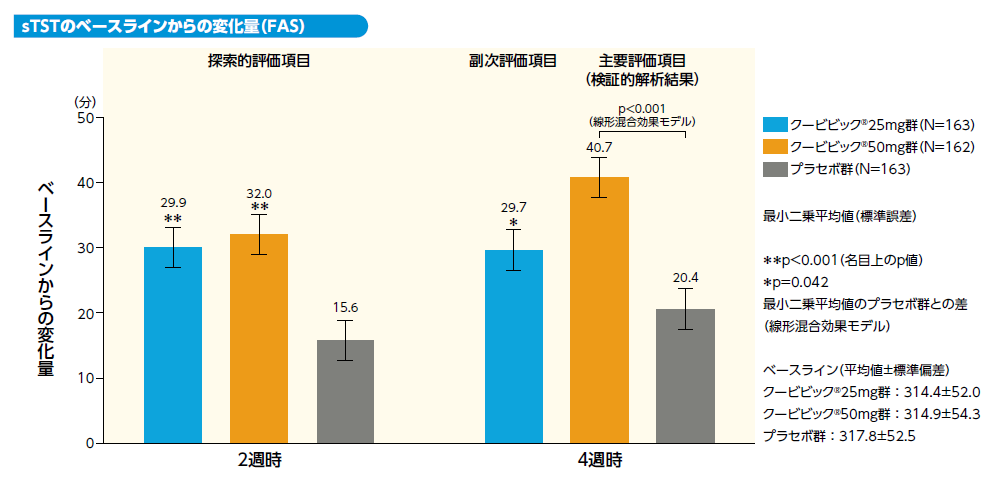
1) 4週時における主観的総睡眠時間(sTST)のベースラインからの変化量[主要評価項目(検証的解析結果)、副次評価項目]
第1の主要評価項目である4週時におけるsTSTのベースラインからの変化量のクービビック50mg群とプラセボ群との差(最小二乗平均値)は20.3分[95%CI:11.4〜29.2]であり、sTSTはプラセボ群と比較してクービビック50mg群で有意に延長しました(p<0.001、線形混合効果モデル)(検証的解析結果)。
副次評価項目である4週時におけるsTSTのベースラインからの変化量のクービビック25mg群とプラセボ群との差(最小二乗平均値)は9.2分[95%CI:0.3〜18.1]であり、 sTSTはプラセボ群と比較してクービビック25mg群で有意に延長しました(p=0.042、線形混合効果モデル)。
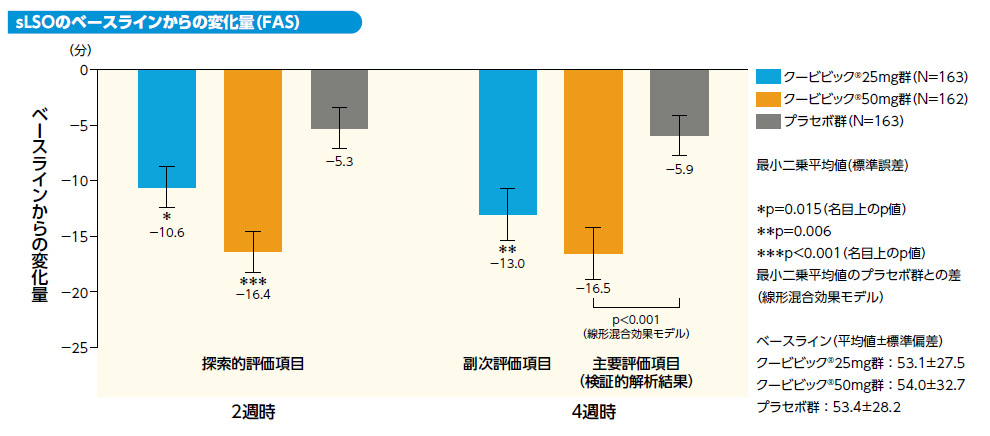
2) 4週時における主観的睡眠潜時(sLSO)のベースラインからの変化量[主要評価項目(検証的解析結果)、副次評価項目]
階層的閉手順により4週時におけるsTSTについてプラセボ群とクービビック50mg群との比較を行い、両側有意水準0.05で有意差が認められたため、4週時におけるsLSOについてプラセボ群とクービビック50mg群を比較しました。
第2の主要評価項目である4週時におけるsLSOのベースラインからの変化量のクービビック50mg群とプラセボ群との差(最小二乗平均値)は−10.7分[95%CI:−15.8〜−5.5]であり、sLSOはプラセボ群と比較してクービビック50mg群で有意に短縮しました(p<0.001、線形混合効果モデル)(検証的解析結果)。
副次評価項目である4週時におけるsLSOのベースラインからの変化量のクービビック25mg群とプラセボ群との差(最小二乗平均値)は−7.2分[95%CI:−12.3〜−2.0]であり、 sLSOはプラセボ群と比較してクービビック25mg群で有意に短縮しました(p=0.006、線形混合効果モデル)。
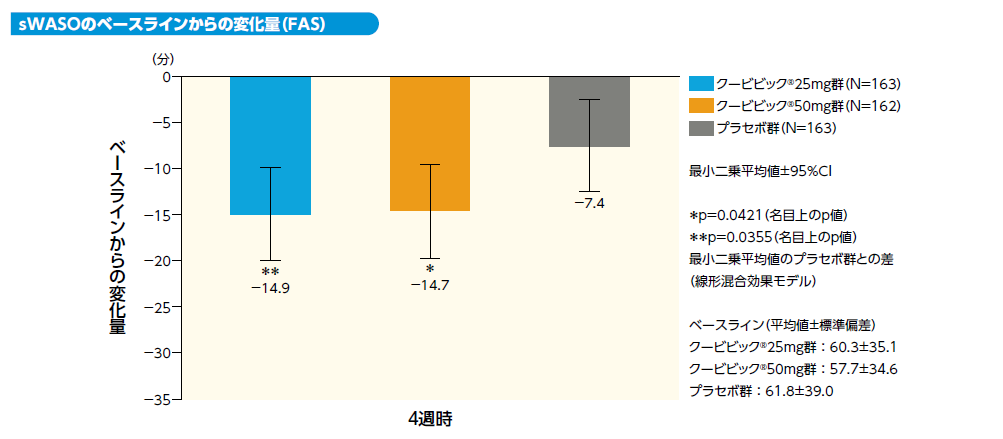
3) 4週時における主観的中途覚醒時間(sWASO)のベースラインからの変化量(事後解析)[探索的評価項目]
本データは事前に計画されていなかったが、承認審査過程で提出し、当局に評価された。不眠症の症状の一つである中途覚醒に対する有効性を理解するうえで重要な情報と判断し掲載する。
4週時におけるsWASOのベースラインからの変化量のクービビック25mg群とプラセボ群との差(最小二乗平均値)は-7.5分[95%CI:-14.6~-0.5]、クービビック50mg群とプラセボ群との差は-7.3分[95%CI:-14.3~-0.3]であり、sWASOはプラセボ群と比較してクービビック25mg群、50mg群で有意に短縮しました(それぞれp=0.0355、p=0.0421、名目上のp値、線形混合効果モデル)。
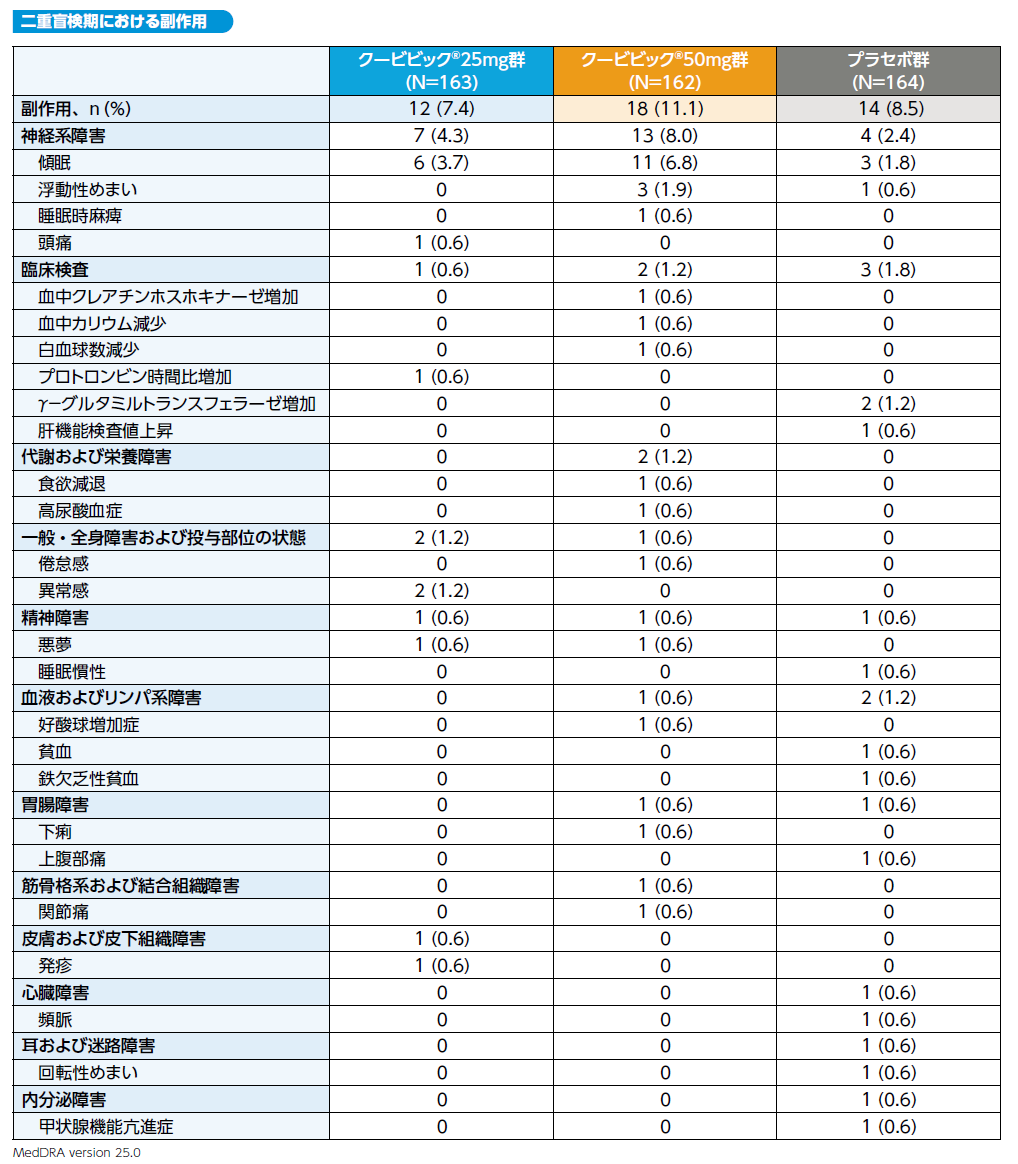
④安全性
1) 有害事象及び副作用
二重盲検期において、有害事象はクービビック25mg群で17.8%(29/163例)、50mg群で22.2%(36/162例)、プラセボ群で 22.6%(37/164例)に、副作用はそれぞれ7.4%(12/163例)、11.1%(18/162例)及び8.5%(14/164例)に発現しました。
2) 重篤な副作用
本試験において、重篤な副作用は報告されませんでした。
3) 投与中止に至った副作用
二重盲検期に投与中止に至った副作用は、クービビック25mg群で1例(0.6%)に発疹、50mg群で1例(0.6%)に浮動性めまいが報告されました。
MedDRA version 25.0
本試験において、死亡例は報告されませんでした。
反跳性不眠は、プラセボrun-out期(来院6回目の前7日間の平均値)におけるsTST及びsLSOのベースラインからの変化量により評価しました。
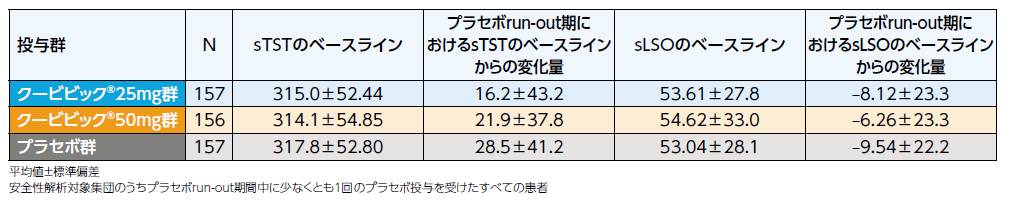
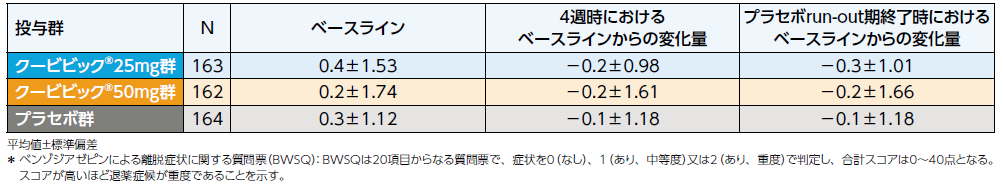
ベンゾジアゼピンによる離脱症状に関する質問票(BWSQ)*合計スコアを用いて退薬症候を評価したところ、プラセボrun-out期終了時におけるベースラインからのスコアの増加の最大値は、クービビック25mg群及び50mg群で1、プラセボ群で7でした。
7) 日中の眠気(JESS)
エプワース眠気尺度日本語版(JESS)*を用いて日中の眠気を評価しました。

8) 翌日の持ち越し効果[WAIS-IV知能検査(符号)]
WAIS-IV知能検査スコア*のベースラインからの変化量を評価しました。
![翌日の持ち越し効果[WAIS-IV知能検査(符号)]](/content/dam/hcp/jp/disease/ncnp/insomnia/quviviq/a304-test/image/a304-test15.png)
8.1 本剤の影響が服用の翌朝以後に及び、眠気、注意力・集中力・反射運動能力等の低下が起こることがあるので、自動車の運転など危険を伴う機械の操作に従事させないよう注意すること。[17.3.1参照]
国内第Ⅲ相長期投与試験(ID-078A305試験)
①試験概要
- 目的
-
日本人不眠症患者におけるクービビック長期投与の安全性を評価する。
- 試験デザイン
-
多施設共同、無作為化、非盲検試験
- 対象
-
不眠症患者154例
<主な登録基準>
・ 18歳以上の男女
・ DSM-5に基づき不眠障害と診断された患者
・ 不眠重症度指数(ISI)スコアが15以上の患者
・ 普段の就床時刻が21:30~00:30、就床時間が6時間から9時間の患者
・ 無作為化(来院2回目)の前7日間に自宅で記入した睡眠日誌において、3夜以上で以下のすべてに該当する患者
(a)主観的睡眠潜時(sLSO)30分以上 (b)主観的中途覚醒時間(sWASO)30分以上 (c)主観的総睡眠時間(sTST)6.5時間以下
・ 睡眠関連呼吸障害(慢性閉塞性肺疾患、睡眠時無呼吸症候群を含む)の既往歴がなく、合併のない患者
・ 周期性四肢運動障害、レストレスレッグス症候群、概日リズム睡眠障害、レム睡眠行動障害、ナルコレプシーをいずれも合併していない患者
・ スクリーニング前12週以内に精神疾患のエピソードがなかった患者 - 方法
-
スクリーニング期(1~4週間)の後、クービビック25mg群又は50mg群に1:2の比で無作為に割付け、非盲検下で治験薬を1日1回就寝前に52週間経口投与した。割付けは、高齢者(65歳以上)、非高齢者を層別因子とした。
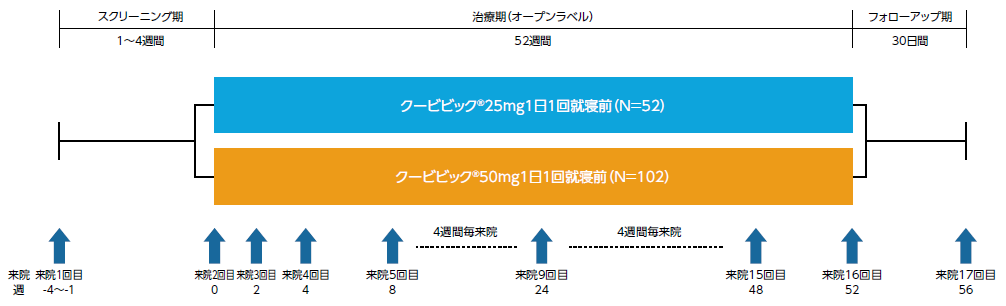
- 評価項目
-
安全性評価項目(主要評価項目):
有害事象、バイタルサイン、臨床検査、心電図、C-SSRS、翌日の持ち越し効果(睡眠日誌のVAS「今朝のあなたの気分について」)、日中の眠気(JESS)等
有効性評価項目(副次評価項目):
・ 以下の項目の各評価時点※1におけるベースライン※2からの変化量
主観的総睡眠時間(sTST)、主観的睡眠潜時(sLSO)、主観的中途覚醒時間(sWASO) 等
※1:評価日及びその直前の6日間における平均値
※2:無作為化(来院2回目)の前7日間に記入した睡眠日誌/不眠症の日中の症状及び影響に関する質問票(IDSIQ)に基づく平均値 - 解析計画
-
解析対象集団:
有効性の解析対象集団は治験薬を1回以上投与されたすべての患者(FAS)、安全性の解析対象集団は治験薬を1回以上投与されたすべての患者とした。
有効性評価項目:
各評価項目の測定値及びベースラインからの変化量について、要約統計量を算出した。評価項目の各評価時点の週平均は、 IDSIQ及びVASは評価日直前の7日間の平均値、それ以外は評価日及びその直前の6日間における平均値とし、 1週間のデータが3日未満であった場合、その週平均は欠測とした。
サブグループ解析:
主観的総睡眠時間(sTST)及び主観的睡眠潜時(sLSO)について、併存疾患(あり、なし)、併存疾患のクラス(精神疾患、精神疾患以外)及び年齢区分(65歳未満、65歳以上)別にサブグループ解析を実施することを事前に計画した。
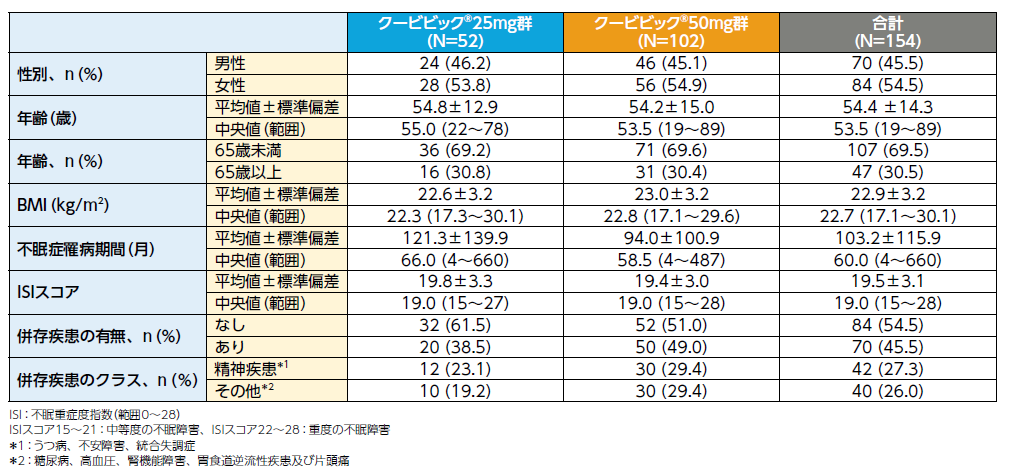
②患者背景(FAS)
③安全性(安全性解析対象集団)(主要評価項目)
有害事象はクービビック25mg群57.7%(30/52例)、50mg群73.5%(75/102例)に、副作用は25mg群19.2%(10/52例)、 50mg群24.5%(25/102例)に発現しました。
主な有害事象(いずれかの投与群で発現頻度5%以上)は、発熱[25mg群17.3%(9/52例)、 50mg群15.7%(16/102例)]、傾眠[25mg群13.5%(7/52例)、50mg群14.7%(15/102例)]、頭痛[25mg群11.5%(6/52例)、50mg群14.7%(15/102例)]、倦怠感[25mg群0%、50mg群8.8%(9/102例)]、上咽頭炎[25mg群5.8%(3/52例)、50mg群2.0%(2/102例)]、主な副作用(いずれかの投与群で発現頻度5%以上)は、傾眠[25mg群11.5%(6/52例)、 50mg群13.7%(14/102例)]、倦怠感[25mg群0%、 50mg群5.9%(6/102例)]でした。
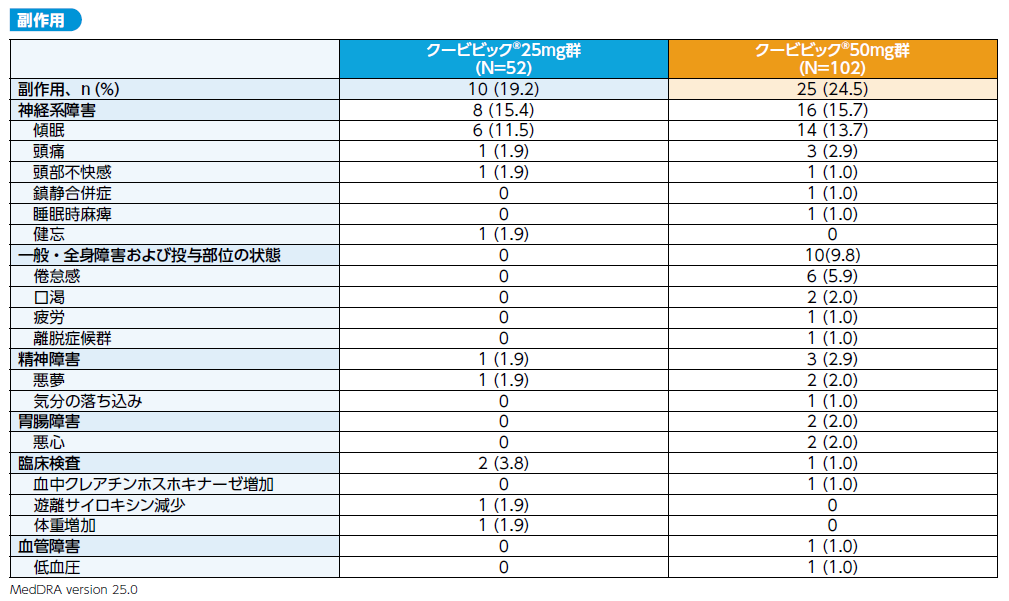
重篤な有害事象は、クービビック25mg群5.8%(3/52例)、50mg群5.9%(6/102例)に認められ、内訳は25mg群で変形性関節症、ギラン・バレー症候群、特発性肺線維症が各1例(1.9%)、50mg群でデュプイトラン拘縮、不安定狭心症、胆管炎、COVID-19、硬膜下血腫、大動脈解離が各1例(1.0%)でした。いずれも治験薬との関連性は否定されました。
3) 投与中止に至った有害事象及び副作用
投与中止に至った有害事象は、クービビック25mg群11.5%(6/52例)、50mg群6.9%(7/102例)に認められ、内訳は25mg群で傾眠2例(3.8%)、ギラン・バレー症候群、COVID-19、妊娠時曝露及び特発性肺線維症が各1例(1.9%)、50mg群で傾眠、倦怠感、頭痛、頚髄神経根障害、COVID-19、コロナウイルス感染、胆管炎及び関節周囲炎が各1例(1.0%)でした。治験薬との関連ありと判定された事象は、25mg群で傾眠2例(3.8%)、50mg群で傾眠、倦怠感及び頭痛が各1例(1.0%)でした。
本試験において、死亡例はありませんでした。
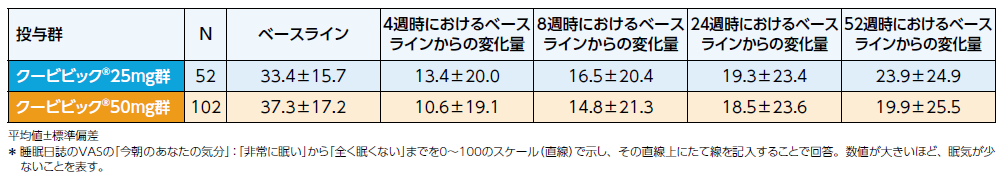
睡眠日誌のVASの「今朝のあなたの気分について」*を測定し、翌日の持ち越し効果を評価しました。
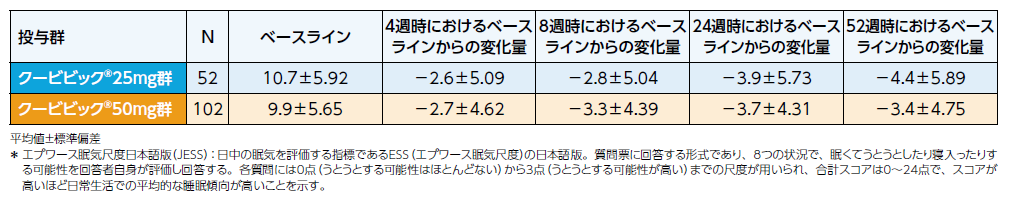
エプワース眠気尺度日本語版(JESS)*を用いて日中の眠気を評価しました。
8.1 本剤の影響が服用の翌朝以後に及び、眠気、注意力・集中力・反射運動能力等の低下が起こることがあるので、自動車の運転など危険を伴う機械の操作に従事させないよう注意すること。[17.3.1参照]
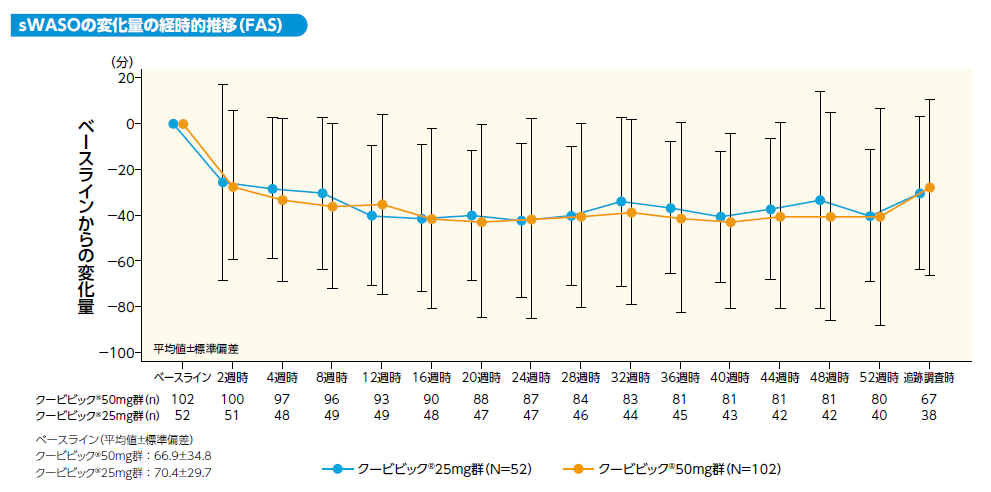
④有効性
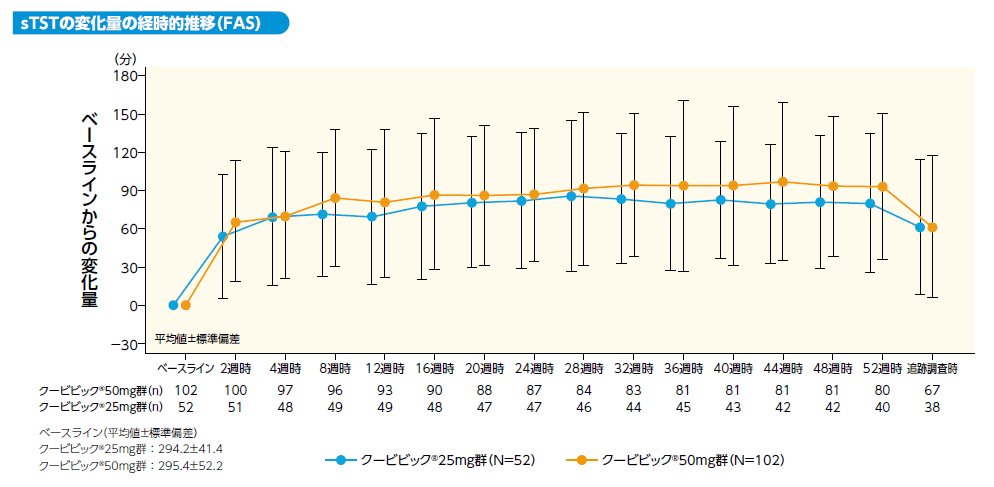
sTSTのベースラインからの変化量は以下のとおりでした。ベースラインからの延長(平均値)はクービビック50mg群で4週時:71分、 8週時:85分、24週時:87分、52週時:94分、クービビック25mg群でそれぞれ70分、72分、82分、81分でした。
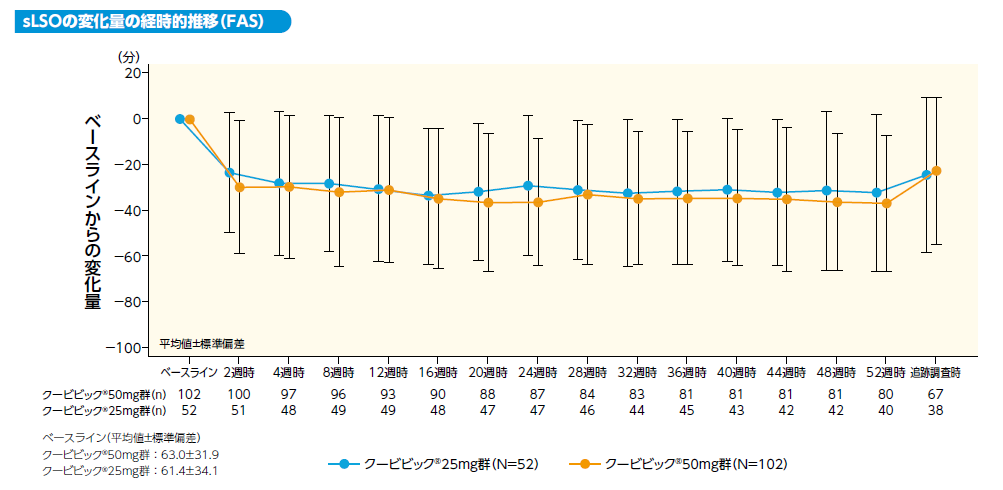
sLSOのベースラインからの変化量は以下のとおりでした。ベースラインからの短縮(平均値)はクービビック50mg群で4週時:29分、 8週時:32分、24週時:36分、52週時:37分、クービビック25mg群でそれぞれ28分、28分、29分、32分でした。
sWASOのベースラインからの変化量は以下のとおりでした。ベースラインからの短縮(平均値)はクービビック50mg群で4週時: 33分、8週時:36分、24週時:41分、52週時:40分、クービビック25mg群でそれぞれ28分、31分、42分、40分でした。